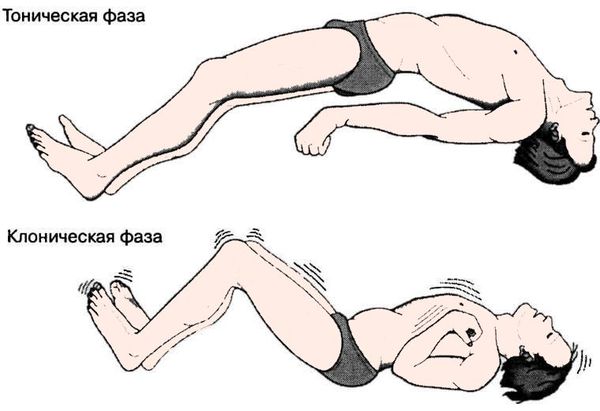
Синдром арнольда киари как причины эпилепсии
Синдром арнольда киари как причины эпилепсии
Синдром арнольда киари как причины эпилепсии
В последнем десятилетии XIX века немецкий патологоанатом Киари описал четыре врожденные аномалии мозжечка и ствола мозга, как проявления нарастающей тяжести единого патогенетического механизма, связанного с давлением сзади на задний мозг при врожденной гидроцефалии.
Основной анатомической особенностью аномалии Киари I типа является разная степень каудального смещения миндалин мозжечка в шейный канал. Аномалия Киари 2 типа характеризуется более тяжелой мозжечковой грыжей, которая, кроме миндалин мозжечка, также включает в себя нижний червь и четвертый желудочек; она, как правило, связана с миеломенингоцеле. Аномалия Киари 3 типа включает в себя признаки высокого менингоэнцефалоцеле с грыжей заднего мозга. Наконец, Киари 4 типа характеризуется тяжелой гипоплазией мозжечка без каудального смещения.
После многих лет и большого количества теоретических, клинических и нейровизуализационных исследований, понимание и осознание этого состояния вышло на новый уровень. В настоящее время считается, что эти четыре порока развития являются отдельными состояниями, с различным патогенезом, клиническим проявлением, и прогнозом, а не разными прогрессирующими стадиями одного заболевания. С анатомической точки зрения аномалии 1-3 типа являются грыжами заднего мозга различной степени, с возможным вторичным образованием полости в спинном мозге, в то время как аномалия 4 типа характеризуется гипоплазией мозжечка. Гипоплазия задней черепной ямки является частой находкой при первых трех аномалиях, но никогда не встречается при четвертой.
У каждой формы свои патогномоничные клинические проявления и по этим причинам различные аномалии Киари должны рассматриваться в разных разделах.
Мальформация Киари 1 типа. Аномалия Киари 1 типа характеризуется неправильным положением (и формой) миндалин мозжечка, которые опускаются из полости черепа ниже большого затылочного отверстия в шейный канал. Это анатомическое нарушение приводит к окклюзии субарахноидального пространства на уровне большого затылочного отверстия с последующим нарушением ликвороциркуляции в головном и спинном мозге; фактически, помимо изменений в задней черепной ямке, повышение ликворного давления на этом уровне приводит к сирингомиелии. Патогенетические механизмы развития аномалии детально изучены и обсуждены в литературе. Различные механизмы патогенеза можно схематично разделить на три категории:
– Гидродинамический (на основе градиента давления между ликворными пространствами головного и спинного мозга).
– Механический (с блоком ликвороциркуляции на уровне большого затылочного отверстия как причины аномалии Киари 1 типа).
– Нарушение развития (с интерпретацией аномалии задней черепной ямки как местного проявления более общего нарушения развития).
Хотя аномалия Киари 1 типа обычно становится симптоматичной в раннем подростковом периоде, в последнее время она обнаруживается с одинаковой частотой как у детей, так и у взрослых.
а) Клиническая картина аномалии Киари I типа. Наиболее частой жалобой у этой группы пациентов является боль в затылке или в шее, усиливающаяся при чихании, кашле или при пробе Вальсальвы. Другими болезненными проявлениями являются боль в плече, спине или боли в конечностях без корешкового распределения. Наиболее частым симптомом являются признаки моторного или сенсорного дефицита в конечностях (> 70%), которые являются проявлением наличия полости в спинном мозге (сирингомиелии). Атаксия тела и конечностей в качестве проявление нарушений в мозжечке является вторым наиболее распространенным симптомом (в 30-40%). Реже (в 15-25%) наблюдается неуклюжесть, нистагм, диплопия, дисфагия и дизартрия как проявление дефицита черепных нервов. Апноэ при заикании наблюдается в 10% случаев, в большинстве случаев проявляясь у младенцев или маленьких детей. Своеобразным проявлением аномалии Киари 1 типа у детей и подростков является прогрессирующий сколиоз (в 30%).
б) Лучевая диагностика. МРТ — лучший способ для диагностики аномалии Киари 1 типа. Важными критериями для этого порока являются: пролабирование одного или обоих миндалин мозжечка ниже большого затылочного отверстия (> 5 мм); возможны шейно-мозговая деформация, отсутствие супратенториальных нарушений (за исключением отдельных случаев небольшого расширения желудочков и обычная локализация четвертого желудочка. Аномалия Киари 1 типа может быть связана с такими костными аномалиями, как маленькая задняя черепная ямка, платибазия, атланто-затылочная ассимиляция, базилярная импрессия, срастание шейных позвонков (аномалия Клиппеля-Фейля). Гидро/сирингомиелия встречается в 50-60% случаев. Она может быть ограничена одной или двумя областями, а может распространяться на всю длину спинного мозга. Обычно полость образуется на уровне С1.
МРТ является полезным дополнением к диагностическим мероприятиям, так как позволяет определить степень сдавления ствола на уровне затылочного отверстия и характеристики ликвородинамики; исследование, выполненное в послеоперационном периоде, позволяет получить некоторое представление об адекватности хирургической декомпрессии.
в) Лечение аномалии Киари I типа. Целью хирургического лечения является декомпрессия задней черепной ямки для восстановления циркуляции спинномозговой жидкости в базальных цистернах и устранение сдавления нервных структур на уровне краниоцервикального перехода. Хирургическое лечение при аномалии Киари 1 типа обычно определяет четко определенный протокол, который включает субокципитальную краниотомию, С1 ламинэктомию, лизис арахноидальных сращений, резекцию миндалин мозжечка (как традиционную тонзилэктомию, так и субпиальную коагуляцию) и расширенную дуропластику. В случае сопутствующей вентральной компрессии (как, например, при платибазии, С1 ассимиляции и т.д.), она должна быть устранена до проведения дорсальной декомпрессии. В последнее время появляются сообщения, которые указывают на возможность достижения сопоставимых результатов при использовании простой декомпрессии, без расширяющейся дуропластики (или с отслоением наружного слоя твердой мозговой оболочки). Интраоперационная ультразвуковая диагностика может быть полезна в этом отношении, демонстрируя синхронизацию движения миндалин с дыханием и сердцебиением, а также наличие адекватного движения ликвора из четвертого желудочка.
Дальнейшие хирургические возможности представлены процедурами, направленными на лечение гидро/сирингомиелии (закупорка задвижки, размещение сиринго-субарахноидального или сиринго-плеврального шунта, и стентирование четвертого желудочка). При сравнении двух основных крупных хирургических вмешательств при лечении аномалии Киари 1 типа с сирингомелией — субокципитальной декомпрессии и установки сиринго-субарахноидального шунта—Hida et al. обнаружили уменьшение размера сирингомиелитической полости у 94% пациентов, перенесших декомпрессию, и у 100% перенесших сирингосубарахноидальное шунтирование. Что касается прогноза, то максимальные преимущества декомпрессии проявляются у пациентов с симптомами, связанными с пароксизмальной внутричерепной гипертензией. Кроме того, пациенты с мозжечковой симптоматикой и синдромом большого затылочного отверстия имеют больше шансов на выздоровление, чем пациенты с центральным синдромом спинного мозга.
При работе с пациентами детского возраста главной проблемой являются показания к операции. Фактически, во многих случаях диагноз аномалии Киари ставится случайно, после МРТ для диагностики причины неспецифических клинических проявлений, таких как головные боли, умственная отсталость, эпилепсия. Большинство авторов не согласно с превентивным хирургическим лечением, считая, что показания к нему должны основываться на наличии симптомов и клинических проявлений, однозначно относящихся к аномалии Киари, а не данными нейровизуализации.
Что касается хирургической техники, некоторые авторы используют те же оперативные приемы, что и для взрослых, т. е. субокципитальную декомпрессию, С1 ламинэктомию, пластику ТМО и резекцию миндалин мозжечка в 40-60% случаев. При послеоперационной МРТ отмечается 100% улучшение симптомов, а также более чем 90% улучшение признаков, связанных с сирингомиелией и уменьшение сирингомиелитических кист в 80% случаев. В последние годы некоторые авторы критикуют этот «классический» подход как слишком тяжелый (а именно интрадуральные манипуляции), особенно в случаях умеренного опущения миндалин. В действительности, по некоторым сообщениям существуют указания на возможность достижения сопоставимых результатов менее инвазивным методом, то есть выполнением субокципитальной декомпрессии (и С1 ламинэктомия) с или без расслаивания внешнего слоя твердой мозговой оболочки. Улучшение или разрешение клинических проявлений описывается у более чем 90% детей, а исчезновение сирингомиелической полости — в 80% случаев. Удаление миндалин мозжечка с вскрытием ТМО или дуропластикой должно использоваться в случаях неэффективности минимально инвазивного лечения.
Аномалия Киари 1 типа.
Сагиттальный срез в Т1-взвешенном режиме МРТ.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Арнольда — Киар
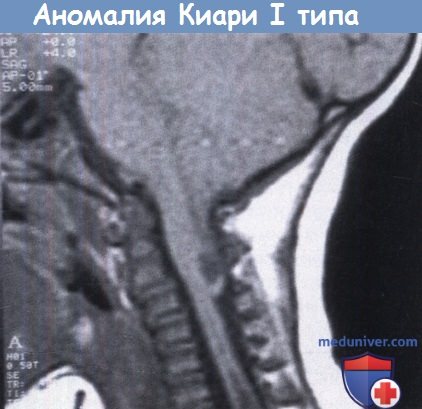
Педиатр Анна Колинько о патологии развития головного мозга, которая может встречаться у 30 % населения
Синдром хронической усталости, головокружения и боль в шее могут быть следствием мальформации (аномалии) Арнольда — Киари. После начала широкого использования МРТ стало понятно, что болезнь встречается у 14–30 % популяции
Мальформация Арнольда — Киари (МАК) — это патология развития ромбовидного мозга: продолговатого и заднего мозга, в последний входит Варолиев мост и мозжечок. При МАК задняя черепная ямка не соответствует мозговым структурам, расположенным в этой области: мозжечок и продолговатый мозг из‑за небольших размеров опускаются ниже большого затылочного отверстия, что приводит к их ущемлению и нарушению ликвородинамики. МАК относят к группе кранио-вертебральных (черепно-позвоночных) мальформаций.
В эпоху до МРТ частота МАК оценивалась от 3,3 до 8,2 наблюдений на 100 000 населения, а у новорожденных — 1 на 4–6 тысяч. Сегодня понятно, что распространенность синдрома Арнольда — Киари значительно больше. Из-за бессимптомного течения и в результате учета разных типов МАК цифры очень разнятся — от 14 до 30 %.
Все первые описания мальформации были посмертными. В 1883 году шотландский анатом Джон Клеланд (J. Cleland, 1835–1925 гг.) впервые описал удлинение ствола и опущение миндалин мозжечка в большое затылочное отверстие у 9 умерших новорожденных. В 1891 году австрийский патолог Ганс фон Киари (H. Chiari, 1851–1916 гг.) подробно охарактеризовал 3 типа мальформации у детей и взрослых. А в 1894 году немецкий патолог Юлиус Арнольд (J. Аrnold, 1835–1915 гг.) подробно описал синдром Киари 2 типа, в сочетании со спинномозговой грыжей (spina bifida). В 1896 году Киари дополнил свою классификацию четвертым типом. В 1907 году ученики Арнольда использовали термин «мальформация Арнольда — Киари» по отношению к аномалии 2 типа. Теперь это название распространилось на все типы. Некоторые врачи справедливо отмечают, что вклад Арнольда несколько преувеличен и верным будет термин «мальформация Киари».
Версии о причинах
Этиология и патогенез синдрома Арнольда — Киари остаются неуточненными. Киари предположил, что смещение мозжечка и продолговатого мозга происходит из‑за внутриэмбриональной гидроцефалии, которая возникает как следствие стеноза сильвиева водопровода — узкого канала длиной 2 см, который соединяет III и IV желудочки мозга.
Клеланд полагал, что аномалия связана с первичным недоразвитием ствола головного мозга. В 1938 г. канадский нейрохирург Уайлдер Пенфилд (W. G. Penfield, 1891–1976 гг.) и его коллега предложили «теорию тяги»: в процессе роста фиксированный спинной мозг втягивает в полость позвоночного канала расположенные выше отделы. В «унифицированной» теории Дэвид Маклон (D. G. McLone) и Пол Неппер (P. A. Knepper) в 1989 году предположили, что первично возникает дефект нервной трубки с истечением ликвора и недостаточным расширением желудочковой системы, что приводит к формированию уменьшенной задней черепной ямки. Однако последующие исследования говорят о том, что существуют разные варианты патологии Арнольда — Киари: с уменьшением задней черепной ямки и без такового, с нарушением ликворооттока и без. Описаны семейные случаи МАК 2 типа, однако роль генетических факторов еще недостаточно изучена.
Типы мальформаций
1 тип — опущение миндалин мозжечка в позвоночный канал ниже уровня большого затылочного отверстия с отсутствием спинномозговой грыжи. У 15–20 % пациентов этот тип сочетается с гидроцефалией, а у 50 % больных — с сирингомиелией — заболеванием, при котором в спинном и продолговатом мозге образуются полости. В 1991 году было предложено подразделить аномалии Арнольда — Киари 1 типа на тип А — с сирингомиелией и тип В — без сирингомиелии.
Сирингомиелии при Арнольде — Киари 1 степени.
Энцефаломенингоцеле — врожденная грыжа головного мозга и его оболочек, содержащая цереброспинальную жидкость.
Спинальная дизрафия — порок развития, заключающийся в отсутствии слияния по средней линии парных закладок кожи, мускулатуры, позвонков, спинного мозга
2 тип — опущение нижних отделов червя мозжечка, продолговатого мозга и IV желудочка. Отличительным признаком данного типа является сочетание со спинномозговой грыжей (spina bifida) в поясничном отделе, отмечается прогрессирующая гидроцефалия, часто — стеноз водопровода мозга. Среди детей с менингомиелоцеле до 90 % случаев сопровождается аномалией Арнольда — Киари 2 степени.
- 3 тип — грубое смещение заднего мозга в позвоночный канал с высокой шейной или подзатылочной грыжей головного мозга и его оболочек и выраженным гипертензивно-гидроцефальным синдромом.
- 4 тип — гипоплазия (недоразвитие) мозжечка без смещения его вниз с эктопией продолговатого мозга.
- 0 тип. В 1998 году американский детский нейрохирург Берманс Искандер (В. J. Iskandar) с коллегами впервые ввел понятие «Киари 0» («Сhiari 0») в описании 5 пациентов, имеющих неврологические симптомы аномалии Арнольда — Киари с сирингомиелией и положением миндалин мозжечка на уровне большого затылочного отверстия. Этот тип так же называют «пограничным с Киари».
0, 1 и 2 степени синдрома Арнольда — Киари наиболее распространены в популяции. III и IV типы обычно несовместимы с жизнью.
Симптоматика
Неврологические симптомы 0 и 1 типов аномалии Арнольда-Киари наиболее часто начинают беспокоить в возрасте 20–40 лет. Степень дислокации миндалин мозжечка может нарастать под влиянием неблагоприятных факторов. Чаще всего жалобами при МАК 0 типа являются головная боль, преимущественно шейно-затылочной локализации, а также боль в шее. Аномалия Арнольда — Киари 1 типа у взрослых чаще проявляется жалобами на нистагм, дизартрию, атаксию, интенционный тремор (тремор при произвольных движениях), головную боль, головокружение, нарушение чувствительности, парезы, нарушение функции тазовых органов, нарушения частоты и ритма пульса, ритма дыхания, лабильность артериального давления, симптомы поражения каудальной группы черепных нервов (IX, X, XI, XII пары) — нарушение чувствительности лица и бульбарные расстройства (расстройства глотания и речи).
Синдром Арнольда-Киари 2 степени впервые проявляется не у взрослых, а у новорожденных или в раннем детском возрасте. МАК 2 типа протекает более тяжело, дети с такой патологией уже рождаются с гидроцефальной формой черепа. Гидроцефалия препятствует нормальному развитию. Кроме того, такие дети страдают нарушениями дыхания, сердцебиения и глотания. Часто заболевание сопровождается судорожными припадками. У детей развивается нистагм, апноэ, стридор, парез голосовых связок, дисфагия с регургитацией, нарушение тонуса в конечностях. Выраженность неврологической симптоматики в первую очередь зависит от выраженности нарушений ликвородинамики, а не от степени эктопии миндалин мозжечка.
Терапия
Лечение аномалий Арнольда — Киари зависит от выраженности неврологической симптоматики. Консервативная терапия включает в себя нестероидные противовоспалительные препараты и миорелаксанты. Если в течение 2–3 месяцев консервативная терапия безрезультатна или у пациента имеется выраженный неврологический дефицит, показано оперативное вмешательство. В процессе операции устраняется сдавление нервных структур и нормализуется ликвороток путем увеличения объема (декомпрессии) задней черепной ямки и установки шунта. Оперативное лечение эффективно, по разным источникам, в 50–85 % случаев, в оставшихся случаях симптоматика регрессирует не полностью. Операцию рекомендуется выполнять до развития тяжелого неврологического дефицита, поскольку восстановление происходит лучше при минимальных изменениях неврологического статуса. Подобное оперативное лечение выполняется почти в каждом федеральном нейрохирургическом центре России и проводится в рамках высокотехнологичной медицинской помощи по системе ОМС.
Пациенты, имеющие мальформацию Арнольда-Киари 0 и 1 типа, могут даже не знать о наличии у себя этого заболевания в течение всей жизни. Вследствие пренатальной диагностики МАК II, III и IV типа дети с данной патологией рождаются всё реже, а современные технологии выхаживания позволяют значительно увеличить продолжительность жизни таких детей.
Лечение аномалии Арнольда-Киари
Госпитализация и лечение по квоте ОМС. Подробнее после просмотра снимков.
Содержание:
Аномалия Арнольда-Киари у взрослых сопровождается опущением и выходом через затылочное отверстие структур головного мозга, которые находятся в задней черепной ямке. При этом находящиеся поблизости мозговые структуры сдавливаются. Это приводит к нарушению оттока цереброспинальной жидкости и развитию гидроцефалии. Чаще всего признаки аномалии Арнольда-Киари сочетаются с сирингомиелией. Это хроническое заболевание центральной нервной системы, которое сопровождается образованием полостей в продолговатом и спинном мозге.
 Неврологи затрудняются с установкой истинных причин развития заболевания. Симптомы аномалии Арнольда-Киари могут возникать из-за уменьшения задней черепной ямки. В результате этого мозговые структуры постепенно выходят через затылочное отверстие. Также причиной может быть увеличение размера мозга на фоне гидроцефалии, черепно-мозговых травм.
Неврологи затрудняются с установкой истинных причин развития заболевания. Симптомы аномалии Арнольда-Киари могут возникать из-за уменьшения задней черепной ямки. В результате этого мозговые структуры постепенно выходят через затылочное отверстие. Также причиной может быть увеличение размера мозга на фоне гидроцефалии, черепно-мозговых травм.
Симптомы заболевания
Симптомы заболевания зависят от типа аномалии Арнольда-Киари. Квалифицировать патологию может невролог или нейрохирург. В клинической практике нейрохирургов часто встречается аномалия Арнольда-Киари 1 степени. Болезнь развивается после окончания периода полового созревания или уже у взрослых пациентов.
Основные признаки патологии у взрослых:
- головная боль в области шеи и затылка, которая склонна к усилению при напряжении мышц, во время кашля и чихания;
- рвота, которая не зависит от приема пищи;
- повышение мышечного тонуса;
- мозжечковая атаксия;
- склонность к нистагму и нарушениям речи;
- снижение остроты зрения и слуха.
Возможно появление и других симптомов, в том числе кратковременной потери сознания. Пациенты с такой болезнью жалуются на головокружение, возникающее всякий раз при повороте головы. В некоторых случаях может даже возникать обморок. Распространенным признаком являются атрофические изменения в области языка и гортани. Они сопровождаются нарушением дыхания и хронической осиплостью голоса. Подобные последствия возникают при длительном отсутствии эффективной медицинской помощи.
Аномалия Арнольда-Киари 2 типа вызывает шумное дыхание, сочетающееся с парезом гортани и забросом пищи в нос. Поставить точный диагноз можно на основании осмотра и данных диагностики. Неврологический осмотр в сочетании с магнитно-резонансной томографией головного мозга и позвоночника позволяет выявить характерные аномалии. При подтверждении диагноза необходимо как можно скорее начинать соответствующее лечение.
Магнитно-резонансная томография является наиболее широко применяемым методом диагностики при данном заболевании. Во время исследования можно обнаружить дислокацию миндалин и получить информацию о составе тканей головного мозга. Для исследования потока цереброспинальной жидкости необходимо проводить фазово-контрастную МРТ.
Аномалия Арнольда-Киари 1 степени
Миндалины мозжечка находятся ниже большого затылочного отверстия. Аномалия Арнольда-Киари 1 степени чаще встречается у взрослых. На фоне развития болезни в центральном канале спинного мозга начинает скапливаться цереброспинальная жидкость. При сочетании болезни с сирингомиелией нарушается чувствительность, появляются признаки онемения и атрофических процессов в мышцах. Возможно исчезновение рефлексов брюшины.
Аномалия Арнольда-Киари 2 степени
При данной болезни через большое затылочное отверстие начинают выходить не только миндалины, но и сам мозжечок, IV желудочек, а также продолговатый мозг. Заболевание связывают с врожденной спинномозговой грыжей. Патология сопровождается миеломенингоцеле, расщеплением позвоночника.
Аномалия Арнольда-Киари 3 и 4 степеней
Аномалия Арнольда-Киари 3-4 степеней сопровождается переходом опустившихся образований мозга в менингоцеле затылка и шеи. При 4 степени болезни гипопластические процессы в мозжечке нередко сочетаются с признаками гидроцефалии.
Возможно развитие сочетанной патологии. Заболевание может возникать на фоне диспластических процессов нервной системы, включая нарушения в функциональности мозолистого тела и недоразвитие подкорковых структур.
Лечение у взрослых
Главным методом лечения является операция. Консервативная терапия может рассматриваться только в качестве дополнительного метода. Но в большинстве случаев заболевание сопровождается значительными неврологическими нарушениями, вплоть до развития парезов и других опасных для жизни осложнений. В таком случае нельзя откладывать хирургическое лечение.
Чаще всего специалисты используют краниовертебральную декомпрессию. Операция предполагает удаление фрагмента кости с расширением затылочного отверстия. Также специалисты ликвидируют признаки сдавления неврологических структур, проводят удаление миндалин мозжечка и частей двух первых шейных позвонков. Для восстановления нормального тока цереброспинальной жидкости используют искусственные материалы и трансплантанты, которые подшивают в область твердой мозговой оболочки в качестве заплаток.
Проведение шунтирующих операций
В нейрохирургии активно используют шунтирующие системы для устранения гидроцефалии. Целью такого хирургического вмешательства является восстановление оттока цереброспинальной жидкости. Шунты имеют односторонние клапаны. В будущем возможно проводить замену этих систем или их ревизию в ходе повторных нейрохирургических вмешательств.
Оттока цереброспинальной жидкости достигают путем дренирования желудочков мозга. Систему шунтирования могут соединять с правым предсердием или брюшной полостью. Возможно применение современных эндоскопических методов оперативного лечения. Они отличаются малоинвазивностью, коротким реабилитационным периодом.
При высокой квалификации нейрохирурга операции проходят без каких-либо осложнений. Риск инфицирования повышается при несоблюдении основных правил инфекционной безопасности, разъединении шунта, резком снижении внутричерепного давления. При использовании специальных шунтирующих систем можно избежать подобных неблагоприятных последствий.
Последствия и прогноз для жизни
При подтверждении диагноза необходимо как можно скорее начинать лечение. Определенные проблемы с диагностикой могут возникать из-за бессимптомного течения болезни. Аномалия Киари 3 степени нередко приводит к летальному исходу. Но своевременно проведенное хирургическое лечение позволяет избежать неблагоприятных последствий. Операция является золотым стандартом современной нейрохирургии.
Даже незначительные неврологические проявления являются показанием для обращения к специалистам. В идеале необходимо начинать лечение сразу после выявления патологии. Болезнь в запущенной форме заканчивается инвалидностью и гибелью пациента.
Своевременно проведенное лечение с использованием операции позволяет избежать необратимых атрофических процессов в центральной нервной системе. В НИИ Бурденко подобные хирургические вмешательства проводят на высоком профессиональном уровне. С пациентами работают опытные нейрохирурги, уделяя повышенное внимание каждому этапу лечения, включая подготовку к операции и проведение тщательного комплексного обследования.
После оперативного вмешательства пациенты находятся под присмотром нейрохирургов, реабилитологов, реаниматологов. Специалисты следят за жизненно важными показателями работы организма. При необходимости профессионалы оказывают первую помощь, тормозят развитие осложнений и других неблагоприятных последствий для здоровья. Современные методики, применяемые в нейрохирургии, позволяет сократить степень повреждения тканей во время работы с головным мозгом, избежать лишних травм и кровопотери.
Консультация нейрохируга
Нейрохирург, доктор медицинских наук
— Нейрохирург 9 отделения НИИ нейрохирургии им. Н.Н.Бурденко (2002 — 2019)
— Ведущий нейрохирург сети клиник «Медси» (2019 — н.в.)
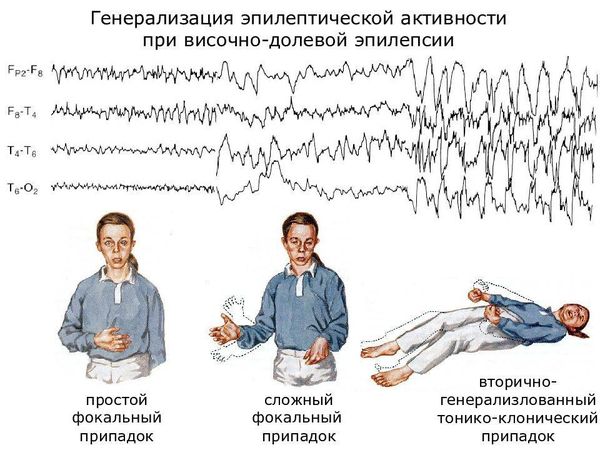
Височная эпилепсия
Височная эпилепсия – особая форма эпилепсии, которая характеризуется локализацией эпилептического очага в височной области головного мозга. Этот тип патологического процесса является одним из наиболее распространенных. Согласно статистике, височная форма эпилепсии составляет порядка 25% случаев этого тяжелого заболевания.
Особенности заболевания
В медицинской практике эпилепсией называют сложное неврологическое заболевание, сопровождающееся внезапными судорожными припадками, приступами. Для болезни характерен тот факт, что в течение приступа человек не осознает, что с ним происходит. В результате пациенты не помнят припадков, не представляют степени их интенсивности и не способны описать, что с ними происходило.
Височная эпилепсия – одна из наиболее распространенных форм заболевания. Этот вид болезни сопровождается парциальными эпилептическими приступами различной степени тяжести. Прогрессирование патологии при отсутствии адекватной терапии грозит возникновением генерализованных припадков, а также развитием различных психических расстройств и деформаций.
Также существует классификации височной эпилепсии, согласно которой выделяют 4 вида патологии, в зависимости от локализации эпилептического очага:
- амигдалярная;
- гиппокампальная;
- латеральная;
- оперкулярная.
Причины возникновения височной эпилепсии
Как и в случае с другими формами заболевания, основной причиной развития височной эпилепсии является формирование в головном мозгу эпилептического очага. В данном случае патологической областью является определенный участок височной доли. Эпилептический припадок провоцируется резким всплеском избыточной активности нейронов в височной доле головного мозга.
Сегодня медики выделяют массу факторов, предрасполагающих развитию височной эпилепсии, разделяя их на две обширные группы:
- Перинатальные – оказывают негативное воздействие на мозг в процессе внутриутробного развития или родовой деятельности. К группе перинатальных факторов относятся различные формы гипоксии плода, травмы, полученные в процессе родов, перенесенные матерью в процессе беременности инфекционные заболевания, асфиксия у новорожденных и т.д.
- Постнатальные – приобретенные факторы постнатального характера гораздо более многочисленны и разнообразны. В числе наиболее распространенных выделяют черепно-мозговые травмы, последствия ишемического или геморрагического инсульта, опухоли мозга, абсцессы, аневризмы и различные нейроинфекции).
Клиническая картина
Основным клиническим признаком височной эпилепсии являются эпилептические припадки. Они возникают внезапно, выражаются в неосознанности пациента, потери ориентации, мышечных судорогах и т.д. При этом характерной особенностью является тот факт, что во время припадка больной поворачивает голову и скашивает глаза к тому виску, в котором сосредоточена область эпилептической активности.
В числе прочих симптомов височной эпилепсии выделяют:
- учащенные, даже систематические головокружения;
- головные боли и мигрени;
- галлюцинации зрительного и слухового типа;
- обонятельные пароксизмы;
- сдавливающее или распирающее ощущение в области сердца;
- нарушения координации;
- различные формы психических отклонений;
- автоматизация жестов и звуков;
- искажения личности и т.д.
Диагностика
Для постановки точного диагноза врач должен сопоставить массу факторов. Первичная диагностика височной эпилепсии включает фиксацию симптомов, частоты приступов, составление подробного анамнеза болезней, выяснение возможных причин или факторов развития заболевания.
На основе полученных в ходе первичной диагностики данных врач определяет необходимые лабораторные и аппаратные исследования:
- МРТ головного мозга;
- ПЭТ КТ;
- электроэнцефалография;
- полисомнография.
Лечение
Полноценного выздоровления пациента, то есть полного отсутствия эпилептических припадков в течение всей жизни добиться крайне тяжело, а в некоторых случаях невозможно. Прибегая к консервативной терапии, главная задача, которую перед собой ставят врачи – свести частоту приступов и прочие проявления заболевания к минимуму.
Консервативное лечение височной эпилепсии предполагает назначение и прием противоэпилепических препаратов.
В течение медикаментозной терапии врач систематически отслеживает состояние пациента, прогресс от приема назначенного лекарства. В случае возникновения нежелательных побочных эффектов лечение корректируется, препарат заменяется или проводится дополнительная симптоматическая терапия.
Если в течение длительного времени положительной динамики от консервативной терапии не наблюдается, может потребоваться хирургическое лечение височной эпилепсии. Для этого привлекаются опытные нейрохирурги, операции при височной эпилепсии проводятся следующие:
- височная резекция;
- фокальная резекция;
- селективная гиппокампотомия;
- амигдалотомия.
В нашей клинике есть все необходимое для точной диагностики, эффективного и комфортного лечения пациентов с эпилептическими приступами. Приём пациентов с приступами потери сознания вдет врач невролог-эпилептолог Палагин Максим Анатольевич.
Наши специалисты

Юшина Мария Александровна
Руководитель центра эпилепсии
и пароксизмальных состояний.
Врач – невролог. Эпилептолог. Озонотерапевт.
Врач – физиотерапевт
Стаж: 7 лет.

Кордонская Ирина Сергеевна
Детский невролог высшей категории.
Эпилептолог.
Нейрофизиолог (врач ЭЭГ – диагностики).
Стаж: 24 года.

Волкова Светлана Анатольевна
Руководитель Центра паркинсонизма и экстрапирамидных заболеваний.
Врач – невролог высшей категории.
Эпилептолог. Озонотерапевт.
Врач – физиотерапевт.
Стаж: 26 лет.

Деревянко Леонид Сергеевич
Руководитель центра диагностики и
лечения нарушений сна.
Врач – невролог высшей категории. Вертебролог. Сомнолог. Эпилептолог. Ботулинотерапевт.
Врач – физиотерапевт.
Стаж: 23 года.

Тарасова Светлана Витальевна
Эксперт № 1 по лечению головной боли и мигрени.
Руководитель центра лечения боли
и рассеянного склероза.
Сомнолог. Эпилептолог. Ботулинотерапевт.
Врач – невролог высшей категории.
Врач – физиотерапевт.
Доктор медицинских наук.
Стаж: 23 года.

Палагин Максим Анатольевич
Врач – невролог. Сомнолог. Эпилептолог. Ботулинотерапевт.
Врач – физиотерапевт.
Стаж: 6 лет.